Structure simulation
Structure simulation functions available in MAISE [1] include: unit
Cell relaxation, Molecular dynamics (MD), and basic Γ-point
Phonon calculations. The structure, the interaction model, and the
job settings are specified in POSCAR, model, and setup
files, respectively. These three files need to be present in the
working directory when running simulations of structures using MAISE.
POSCARcontains a VASP-format representation of the structure.modelcontains information about the interatomic potential.setupcontains specifications on the MAISE run settings.
Cell relaxation
Description
Cell relaxation, also commonly referred to as ‘local structure optimization’, can be performed by using NN or other classical interatomic interaction models available in MAISE. Structure optimizations use analytic derivative-based BFGS [2] or CG [3] algorithms from GSL package. The local optimization is carried out until the maximum number of iterations, MITR, or the targeted enthalpy difference between successive steps, ETOL, is reached.
The unit cell parameters, total atomic energies, and force/stress
components can be outputted at each relaxation step in an OUTCAR
file. The final relaxed structure is saved in a CONTCAR file
containing atomic positions and, optionally, velocities from the last
ionic step of the relaxation. These files are written in the
VASP-style format which can be utilized by external codes to perform
vibrational property analysis, global structure optimization, etc.
Table 1 lists setup parameters relevent for cell
relaxation simulations using MAISE.
FLAG |
Short Description |
|---|---|
job type: structure relaxation (20) |
|
number of cores for parallel run |
|
dimensionality of unit cell: crystal (3); cluster (0) |
|
maximum number of cell optimization steps |
|
error tolerance for cell optimization convergence |
|
cell optimization type: force only (2); full cell (3); volume (7) |
|
external pressure in GPa |
|
output options in OUTCAR file |
|
minimizer type: BFGS2 (0); CG-FR (1); CG-PR (2); steepest descent (3) |
|
strength of the additional repulsion in NN potentials |
|
fraction of shortest bond to Rmin to consider structure collapsed |
JOBT controls the job type. It should be set to 20 for structure relaxations.
NPAR sets the number of cores that will be allocated toward processing the MAISE run. This task is internally parallelized over the number of atoms, so NPAR should not exceed the total number of atoms in the system. Ideally, for equal distribution of load, it is recommended that number of atoms in the system be a multiple of NPAR.
NDIM controls whether the system that is being relaxed is treated as a periodic boundary structure type (wire, film, bulk) or of a cluster type (nanoparticle). Failure to define NDIM properly will yield unphysical results.
3 for periodic boundary structure types
0 for cluster structure types
MITR controls the maximum number of optimization steps. If the error tolerance described in ETOL is reached sooner, the relaxation will stop regardless of MITR.
ETOL controls error tolerance for the optimization convergence. If the difference in the total enthalpy in eV between two successive steps never falls below this value the relaxation will run to MITR steps.
RLXT controls the cell optimization type. It is similar to the ISIF tag in VASP. Currently allowed values are:
Value |
Degree of freedom |
||
|---|---|---|---|
atomic position |
cell shape |
cell volume |
|
2 |
yes |
no |
no |
3 |
yes |
yes |
yes |
7 |
no |
no |
yes |
PGPA controls the external hydrostatic pressure in GPa that is being applied to the system.
COUT controls how much infromation is outputted in the OUTCAR file regarding the energy and forces of the system.
Value |
Energy |
Force |
|---|---|---|
00 |
final step |
no |
01 |
first and final steps |
no |
02 |
all steps |
no |
10 |
final step |
final step |
11 |
first and final steps |
first and final steps |
12 |
all steps |
all steps |
MINT controls which analytic derivative-based algorithm from the GSL package is used for relaxation. Full documentation for these algorithms can be found at this URL. Currently MAISE has the following options:
0 BFGS2: recommended version of Broyden–Fletcher–Goldfarb–Shanno algorithm
1 CG-FR: Fletcher-Reeves conjugate gradient algorithm
2 CG-PR: Polak-Ribiere conjugate gradient algorithm
3 the steepest descent algorithm
Note
Presently, MAISE enforces symmetry only for unit cells with orthogonal lattice vectors by setting to zero all off-diagonal stresses below \(10^{-8}\) eV/A \(^{3}\) and atomic force components below \(10^{-8}\) eV/A. For example, the in-plane symmetry of hexagonal unit cells might not be preserved during full structural relaxations.
UREP defines the strength of an additional repulsive term on top of a NN potential when interatomic distances fall below a minimum value that depends on the elements. In practically all cases, use the default of 100.0 eV/Ang. To switch it off, set it to a negative value.
FRAC defines the ratio of the interatomic distance to an element-dependent minimum distance allowed between two atoms. If the ratio falls below the specified value the relaxation prints out an ERROR message in the OUTCAR file. In most cases, use the default value of 1.0. It is set to a lower value (e.g., 0.95) only during EVOS runs generating NN reference data, as this enables the addition of ‘slightly collapsed’ configurations.
Examples
The following example displays the relaxation of a slighlty distorted
FCC-Ag structureusing our most recent NN interatomic potential
model. In order to run a cell relaxation, the working directory
should include the following files: maise, setup, model,
and POSCAR.
The setup file should be adjusted using the FLAG settings
described above under Cell relaxation. The settings used in this
example are:
======================================================================================
JOB/MODEL types
======================================================================================
JOBT 20
======================================================================================
OPTIMIZATION parameters
======================================================================================
NPAR 4
NDIM 3
MITR 30
ETOL 1e-8
RLXT 3
PGPA 0.0
COUT 12
MINT 0
The starting POSCAR structure before relaxation used in this
example is displayed below.
Ag
1.00000000000000
4.16409010773519 0.00000000000000 0.00000000000000
0.00000000000000 4.16409010773519 0.00000000000000
0.00000000000000 0.00000000000000 4.16409010773519
Ag
4
direct
0.00000000000000 0.00000000000000 0.00000000000000
0.00000000000000 0.50020000000000 0.50000000000000
0.50000000000000 0.00000000000000 0.50000000000000
0.50000000000000 0.50000000000000 0.00000000000000
In order to relax this structure, run the executable maise.
$ maise
The output printed out on the screen in this case is:
=======================================================================
| Module for Ab Initio Structure Evolution |
| version maise.2.7.00 |
| https://github.com/maise-guide/maise |
=======================================================================
| Cell relaxation |
=======================================================================
BFGS2 relaxation: 21 adjustable parameters
0 -11.3100852801119967
1 -11.3100866766688579 -0.0000013965568613
2 -11.3100868572737170 -0.0000001806048591
3 -11.3100868572736903 0.0000000000000266
ENERGY CALLS = 15
FORCE CALLS = 3
INI -11.31008528
DIF -0.00000158
FIN -11.31008686
After a successful relaxation, the following three files are produced
in the working directory: CONTCAR, OSZICAR, and OUTCAR.
CONTCAR provides the relaxed structure.
Ag
1.00000000000000
4.16409012368350 0.00000000000000 0.00000000000000
0.00000000000000 4.16409012591455 0.00000000000000
0.00000000000000 0.00000000000000 4.16409012368350
Ag
4
direct
0.00000000000000 0.00004999999182 0.00000000000000
0.00000000000000 0.50005000007339 0.50000000000000
0.50000000000000 0.00004999999182 0.50000000000000
0.50000000000000 0.50004999994298 0.00000000000000
OSZICAR contains a list of enthalpies at each iterated relaxation step of the run.
-11.31008528
-11.31008668
-11.31008686
-11.31008686
OUTCAR is a summary of the run, with its content dependent on the COUT FLAG.
----------------------------------------------------------------------------------------------------------
Cell optimization with maise.2.5.00 model 00D7FB00 on Thu Jul 9 10:05:18 2020
----------------------------------------------------------------------------------------------------------
optimizer type 0
relaxation type 3
target pressure 0.00 GPa
dimensionality 3
species Ag
atoms of each species 4
total number of atoms 4
----------------------------------------------------------------------------------------------------------
----------------------------------------------------------------------------------------------------------
LATTICE VECTORS VOLUME (Angst^3/atom)
----------------------------------------------------------------------------------------------------------
4.164090 0.000000 0.000000 18.050963 vol
0.000000 4.164090 0.000000
0.000000 0.000000 4.164090
----------------------------------------------------------------------------------------------------------
POSITION (Angst) TOTAL-FORCE (eV/Angst) ATOM ENERGY (eV)
----------------------------------------------------------------------------------------------------------
0.000000 0.000000 0.000000 0.000000 0.001961 0.000000 -2.827521316230
0.000000 2.082878 2.082045 0.000000 -0.003788 0.000000 -2.827520865463
2.082045 0.000000 2.082045 0.000000 0.001961 0.000000 -2.827521316230
2.082045 2.082045 0.000000 0.000000 -0.000134 0.000000 -2.827521782188
----------------------------------------------------------------------------------------------------------
Total 0.00000010 0.00000011 0.00000010 0.00000000 0.00000000 0.00000000
in kB 0.00015907 0.00017858 0.00015907 0.00000000 0.00000000 0.00000000
----------------------------------------------------------------------------------------------------------
iter 0 total enthalpy= -11.31008528 energy= -11.31008528 -2.82752132 -2.82752132
----------------------------------------------------------------------------------------------------------
----------------------------------------------------------------------------------------------------------
LATTICE VECTORS VOLUME (Angst^3/atom)
----------------------------------------------------------------------------------------------------------
4.164090 0.000000 0.000000 18.050963 vol
0.000000 4.164090 0.000000
0.000000 0.000000 4.164090
----------------------------------------------------------------------------------------------------------
POSITION (Angst) TOTAL-FORCE (eV/Angst) ATOM ENERGY (eV)
----------------------------------------------------------------------------------------------------------
0.000000 0.000248 0.000000 0.000000 -0.000378 0.000000 -2.827521668417
0.000000 2.082398 2.082045 0.000000 -0.000434 0.000000 -2.827521709198
2.082045 0.000248 2.082045 0.000000 -0.000378 0.000000 -2.827521668417
2.082045 2.082028 0.000000 0.000000 0.001190 0.000000 -2.827521630637
----------------------------------------------------------------------------------------------------------
Total 0.00000001 0.00000001 0.00000001 0.00000000 0.00000000 0.00000000
in kB 0.00001465 0.00001876 0.00001465 0.00000000 0.00000000 0.00000000
----------------------------------------------------------------------------------------------------------
iter 1 total enthalpy= -11.31008668 energy= -11.31008668 -2.82752167 -2.82752167
----------------------------------------------------------------------------------------------------------
----------------------------------------------------------------------------------------------------------
LATTICE VECTORS VOLUME (Angst^3/atom)
----------------------------------------------------------------------------------------------------------
4.164090 0.000000 0.000000 18.050963 vol
0.000000 4.164090 0.000000
0.000000 0.000000 4.164090
----------------------------------------------------------------------------------------------------------
POSITION (Angst) TOTAL-FORCE (eV/Angst) ATOM ENERGY (eV)
----------------------------------------------------------------------------------------------------------
0.000000 0.000208 0.000000 0.000000 0.000000 0.000000 -2.827521714318
0.000000 2.082253 2.082045 0.000000 0.000000 0.000000 -2.827521714318
2.082045 0.000208 2.082045 0.000000 0.000000 0.000000 -2.827521714318
2.082045 2.082253 0.000000 0.000000 0.000000 0.000000 -2.827521714318
----------------------------------------------------------------------------------------------------------
Total -0.00000000 -0.00000000 -0.00000000 0.00000000 0.00000000 0.00000000
in kB -0.00000343 -0.00000358 -0.00000343 0.00000000 0.00000000 0.00000000
----------------------------------------------------------------------------------------------------------
iter 2 total enthalpy= -11.31008686 energy= -11.31008686 -2.82752171 -2.82752171
----------------------------------------------------------------------------------------------------------
----------------------------------------------------------------------------------------------------------
LATTICE VECTORS VOLUME (Angst^3/atom)
----------------------------------------------------------------------------------------------------------
4.164090 0.000000 0.000000 18.050963 vol
0.000000 4.164090 0.000000
0.000000 0.000000 4.164090
----------------------------------------------------------------------------------------------------------
POSITION (Angst) TOTAL-FORCE (eV/Angst) ATOM ENERGY (eV)
----------------------------------------------------------------------------------------------------------
0.000000 0.000208 0.000000 0.000000 0.000000 0.000000 -2.827521714318
0.000000 2.082253 2.082045 0.000000 0.000000 0.000000 -2.827521714318
2.082045 0.000208 2.082045 0.000000 0.000000 0.000000 -2.827521714318
2.082045 2.082253 0.000000 0.000000 0.000000 0.000000 -2.827521714318
----------------------------------------------------------------------------------------------------------
Total -0.00000000 -0.00000000 -0.00000000 0.00000000 0.00000000 0.00000000
in kB -0.00000343 -0.00000358 -0.00000343 0.00000000 0.00000000 0.00000000
----------------------------------------------------------------------------------------------------------
iter 3 total enthalpy= -11.31008686 energy= -11.31008686 -2.82752171 -2.82752171
----------------------------------------------------------------------------------------------------------
Total CPU time used (sec): 0.57613000 wall time (sec): 0.12232300
For more examples, please view examples of relaxation directory on MAISE’s Github page.
Molecular dynamics
Description
MD simulations can be run in the microcanonical ensemble (NVE) with the Verlet algorithm [4],
the canonical ensemble (NVT) with the Nosé-Hoover thermostat [5][6]a and isobaric-
isothermal ensemble (NPT) with a combination of the Nosé-Hoover thermostat and the Berend-
sen barostat [7]. The velocities are initialized either according to the Maxwell distribution at
a given starting temperature or with the values specified in the POSCAR file. MAISE outputs
energies, lattice parameters, the Lindemann index, average RDF, etc. for each temperature. In the current version of MAISE, the Lindemann index value is well-defined only for NPs and the barostat is implemented for unit
cells with orthogonal lattice vectors.
Table 2 lists setup parameters relevant for MD simulations.
FLAG |
SHORT DESCRIPTION |
|---|---|
job type: molecular dynamics (21) |
|
MD run type: NVE (10); NVT (20); NPT (30); isobaric-isothermal (40) |
|
number of cores for parallel run |
|
starting temperature of the simulation |
|
final temperature of the simulation |
|
temperature increment during the simulation |
|
integration timestep in femtoseconds |
|
number of integration steps per temperature |
|
thermostat coupling constant |
|
barostat coupling constant |
|
isothermal compressibility in 1/GPa |
|
dimentionality of the unit cell: crystal (3); cluster (0) |
JOBT controls the job type. It should be set to 21 for molecular dynamics.
MDTP controls MD run type. Currently we allow following types of MD runs:
10 NVE run with the Verlet algorithm
20 NVT run with the Nosé-Hoover thermostat
30 NPT run with the combination of the Nosé-Hoover thermostat and the Berendsen barostat.
40 allow the system to evolve in the isobaric-isothermal ensemble
Note
If the user wishes to provide custom starting velocities as opposed
to the Maxwell distribution, the MDTP flage should be
increased by 1. For example, to define starting velocities and
perform an NVT run, please include velocities at the end of
POSCAR and change MDTP from 20 to 21.
NPAR sets the number of cores that will be allocated toward processing the MAISE run. This task is internally parallelized over the number of atoms, so NPAR should not exceed the total number of atoms in the system. Ideally, for equal distribution of load, it is recommended that number of atoms in the system be a multiple of NPAR.
TMIN controls the initial temperature of the simulation.
TMAX controls the final temperature of the simulation.
TSTP controls the temperature increment during the simulation. If TMIN is equal to TMAX the value of TSTP is irrelevant.
DELT defines the value of the integration time step in femtoseconds. Typical values are 1-2 fs.
NSTP controls how many integration steps are carried out per each temperature step.
CPLT controls coupling constant for thermostat. Its values should lie within the range 0-100. Lower values for this parameter correspond to a more loosely coupled system with the heat bath. If the user does not provide this parameter, it is defaulted to 25.
CPLP controls coupling constant for barostat. Its values should lie within the range 0-100. Lower values for this parameter correspond to a more loosely coupled system with the pressure bath. If the user does not provide this parameter, it is defaulted to 50.
ICMP controls isothermal compressibility for the system. This parameter is relevant in runs when barostat is present.
NDIM controls whether the simulated structure is treated as periodic (wire, film, bulk) or non-periodic (nanoparticle). Failure to define NDIM properly will yield unphysical results.
3 for periodic boundary structure types
0 for cluster structure types
Examples
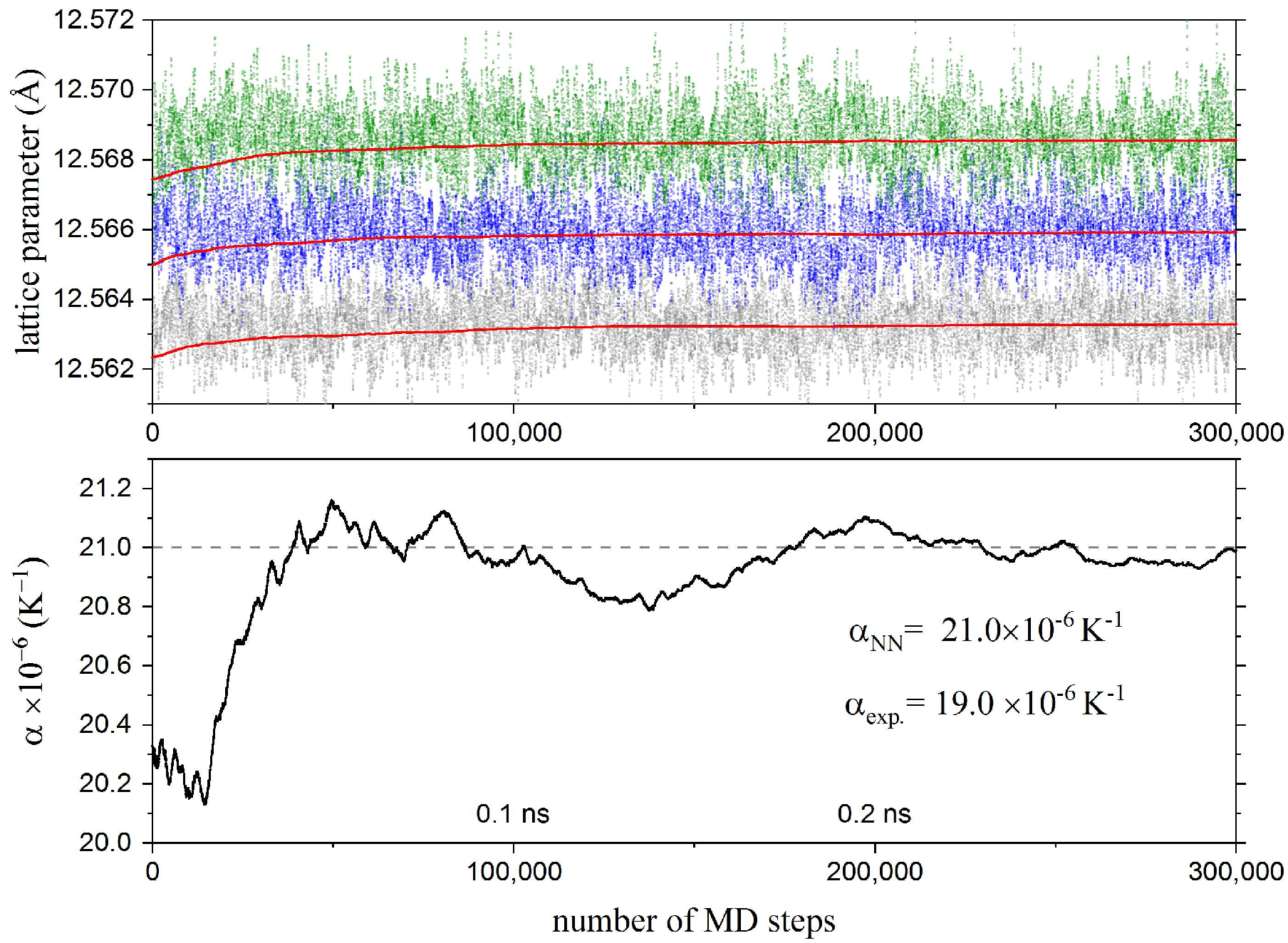
In the example featured in Ref. [1], the linear thermal expansion coefficients around 300K were calculated for 3 elements: Ag, Cu, and Na.
To achieve this, NPT runs were carried out at temperatures 290K, 300K and 310K. The linear expansion coefficients were calculated by numerically differentiating the lattice constants with respect to the temperature.
To run each of the calculations, the following 4 files need to be present in the working directory: maise, POSCAR, setup, and model.
POSCARcontains a representation of the structure.modelis an interatomic interaction potential model relevant to the system.setupcontains the FLAG settings for the MAISE run.
For these calculations the following settings were specified in the setup file.
===============================================================================
JOB/MODEL types
===============================================================================
JOBT 21
===============================================================================
NPAR 16
PGPA 0.0
===============================================================================
MOLECULAR DYNAMICS parameters
===============================================================================
MDTP 30
TMIN 290.0
TMAX 290.0
TSTP 20.0
DELT 1.0
NSTP 500000
CPLT 25.0
CPLP 100.0
ICMP 0.007
NDIM 3
Each run is carried out by executing MAISE:
$ maise
After a successful run, several files listed below will be produced in the working directory.
ave-out.datcontains the final averages for potential energy, kinetic energy, internal pressure, lattice parameter, change in energy, and Lindemann index.AVERAGE-TTTis the “average” structure for the given temperature TTT - a structure created by averaging atomic positions.CONTCARis the final structure at the end of the run.CONTCAR-TTTis the structure at the end of the temperature TTT.RDF-TTT.datis the average radial distribution function (RDF) at temperature TTT.TEMP-TTT.datcontains intermediate values (at every 10 integration steps) of the parameters present inave-out.dat.
The following plot shows the lattice parameters during the MD runs and the convergence of the linear thermal expansion coefficient for Ag.
Phonon calculations
Description
MAISE can perform basic phonon calculations by evaluating the atomic forces for a given relaxed structure using a NN interatomic model or a traditional potential available in MAISE. Our studies of vibrational properties [8] have been performed with an external PHON package [9] because it readily links with VASP or MAISE for a consistent comparison of the NN models against the DFT.
Presently, MAISE has an internal option to calculate Γ-point phonons with the frozen phonon method in the quasi-harmonic approximation. The dynamical matrix is constructed by numerical differentiation of the atomic forces. The magnitude of atomic displacements of each atom is defined by the DISP parameter. Due to the negligible numerical noise of the NN analytic forces, the displacement values can be kept small to reduce the anharmonic effects and satisfy the acoustic sum rule.
The main application of this basic feature is to determine the
presence of soft frequencies in the analysis of structures’ dynamical
stability. The code marks trivial zero-frequency translational (and
rotational) modes by checking whether the eigenvectors generate net
linear (and angular) momenta in crystals (and clusters). Ordered
frequencies and the corresponding eigenvectors are printed in the
OUTCAR file and can be used for introducing soft-mode mutations in
global evolutionary searches [10] or monitoring nudged elastic band
method convergence in transition state searches.
Table 3 lists setup parameters relevent for phonon calculation simulations using MAISE.
FLAG |
SHORT DESCRIPTION |
|---|---|
job type: phonon calculations (22) |
|
size of the displacement made to each atom in Angstroms |
|
number of cores for parallel run |
|
dimentionality of the unit cell: crystal (3); cluster (0) |
JOBT controls the job type. It should be set to 22 for phonon calculations.
DISP is the size of the displacement made to each atom in Angstroms. If no value is provided it is defaulted to 0.001. It can be kept small because of the negligible numerical noise in the calculation of atomic forces as analytic derivatives.
NPAR sets the number of cores that will be allocated toward processing the MAISE run. This task is internally parallelized over the number of atoms, so NPAR should not exceed the total number of atoms in the system. Ideally, for equal distribution of load, it is recommended that number of atoms in the system be a multiple of NPAR.
NDIM controls whether the simulated structure is treated as periodic (wire, film, bulk) or non-periodic (nanoparticle). Failure to define NDIM properly will yield unphysical results.
3 for periodic boundary structure types
0 for cluster structure types
Examples
In the following example a Γ-point phonon calculation is carried out for a CuAg system.
Initially, only the files listed below are needed in the working
directory for the phonon calculation: maise, POSCAR,
model, and setup.
POSCARcontains a representation of the structure. In this example, CuAg is a cubic unit cell.modelis an interatomic interaction potential model relevant to the system. This example uses CuAg.setupcontains the FLAG settings for the MAISE run.
The setup file should contain the following settings:
=========================================================================================
JOB/MODEL types
=========================================================================================
JOBT 22
=========================================================================================
OPTIMIZATION parameters
=========================================================================================
DISP 0.001
NPAR 12
ETOL 1e-8
COUT 12
NDIM 3
=========================================================================================
The POSCAR file used in this example is:
CuAg
1.00000000000000
6.23344492208666 0.00000000000000 0.00000000000000
0.00000000000000 6.23344492208668 0.00000000000000
0.00000000000000 0.00000000000000 3.11672246104334
Cu Ag
4 4
direct
0.00000000000000 0.00000000000000 0.00000000000000
0.00000000000000 0.50000000000000 0.00000000000000
0.50000000000000 0.00000000000000 0.00000000000000
0.50000000000000 0.50000000000000 0.00000000000000
0.25000000000000 0.25000000000000 0.50000000000000
0.75000000000000 0.25000000000000 0.50000000000000
0.25000000000000 0.75000000000000 0.50000000000000
0.75000000000000 0.75000000000000 0.50000000000000
In order to calculate phonons, run MAISE:
$ maise
The output produced should resemble the following:
=======================================================================
| Module for Ab Initio Structure Evolution |
| version maise.2.7.00 |
| https://github.com/maise-guide/maise |
=======================================================================
| Phonon calculation |
=======================================================================
Eigenvalues at the Gamma point (cm-1)
0 190.1189703392989543
1 190.1189703392320780
2 179.0697927772321236
3 179.0697927772046967
4 179.0697916398272014
5 148.9589198226848339
6 147.2041327277512721
7 147.2041327277389939
8 147.2041314956150586
9 147.2041314956086353
10 141.2417901193579155
11 141.2417901192900445
12 136.6290152004455649
13 136.6290152004243623
14 108.8997952619890128
15 108.5283917347479274
16 108.5283917347334182
17 108.5283907788708575
18 108.5283907788655426
19 0.0201311023078189
20 0.0201305313694524
21 0.0198312735576416
22 -4.7239924737495516
23 -4.7239924776967381
For this periodic structure, MAISE highlighted the eigenvalues corresponding to the trivial translations and determined two degenerate imaginary modes.
After a successful MAISE run, the following files will be produced in the working directory: OUTCAR, OSZICAR, and CONTCAR.
OUTCARis the main output file with all the eigenvalues and eigenvectors. For instance, eigenmode 22 has the following atomic displacements:
eigenvalue 22 -4.7239924737495516 (cm-1)
0 -0.3326247221074291 0.0968110110123686 0.0000000000022677
1 0.3326247221511021 -0.0968110108370395 0.0000000000022681
2 0.3326247221510530 -0.0968110108369316 0.0000000000022679
3 -0.3326247221076773 0.0968110110120847 0.0000000000022679
4 0.1007547475905676 -0.3461746714541514 0.0000000000031000
5 -0.1007547475342395 0.3461746716829493 0.0000000000030273
6 -0.1007547475334858 0.3461746716842512 0.0000000000031703
7 0.1007547475904161 -0.3461746714542330 0.0000000000025346